INTRODUCTION
Inflammatory bowel disease (IBD), including CD and UC, is a chronic, intestinal inflammatory disease that is often associated with the development of various extraintestinal symptoms. Extraintestinal symptoms can be divided into 2 groups: extraintestinal manifestations (EIMs) and extraintestinal complications. EIMs are defined in patients with IBD when an inflammatory pathology is located outside the intestine, the pathogenesis of which depends on the extension/translocation of immune pathways from the intestine, is an independent inflammatory event sustained by IBD, or shares a common environmental or genetic predisposition with IBD [1]. EIMs can be divided into 3 categories: EIMs directly related to bowel disease activity, including episcleritis, erythema nodosum (EN), oral aphthous ulcers, and pauciarthritis; those not related to bowel disease activity but simply reflecting the patient’s susceptibility to related autoimmune disorders, including ankylosing spondylitis (AS) and uveitis; and those with an unclear relationship to bowel disease activity, including pyoderma gangrenosum (PG) and primary sclerosing cholangitis (PSC). Extraintestinal complications are initiated by the disease itself and consequently include micronutrient deficiency, osteoporosis, peripheral neuropathy, nephrolithiasis, and IBD drug-related side effects [2]. EIMs or extraintestinal complications occur in various organs and significantly affect the quality of life of patients with IBD (Table 1). Physicians who treat IBD need to keep track of the progress and treatment of these EIMs or extraintestinal complications of the disease through multidisciplinary approaches. Herein, we will focus on and review the pathogenesis and clinical perspectives of EIMs in IBD.
WHY ARE EIMs IMPORTANT IN IBD?
The prevalence of EIMs is strongly influenced by the age at diagnosis and sex [3,4] and is reported to be 23.0% in CD and 24.4% in UC, according to data from the National Health Insurance System of Korea [3]. Up to 50% of patients with IBD experience at least one EIMs, which can develop even before the diagnosis of IBD [5]. However, these results of EIMs differ greatly among study methods and research designs.
Abdominal pain is a common symptom related to IBD, and pain is the principal symptom in IBD [6]. Pain is a longstanding problem and can also be seen in EIMs [7]. During disease flares, pain is present in 50%-70% of patients with IBD, and the health-related quality of life (HRQOL) of these patients was reported to be significantly lower in patients suffering from pain than in those without pain [6,7].
As such, EIMs are a common problem and may significantly impact the quality of life of patients with IBD, requiring specific treatments depending on the affected organs. Therefore, successful management of EIMs is essential for improving the quality of life of patients with IBD.
PATHOGENESIS OF EIMs
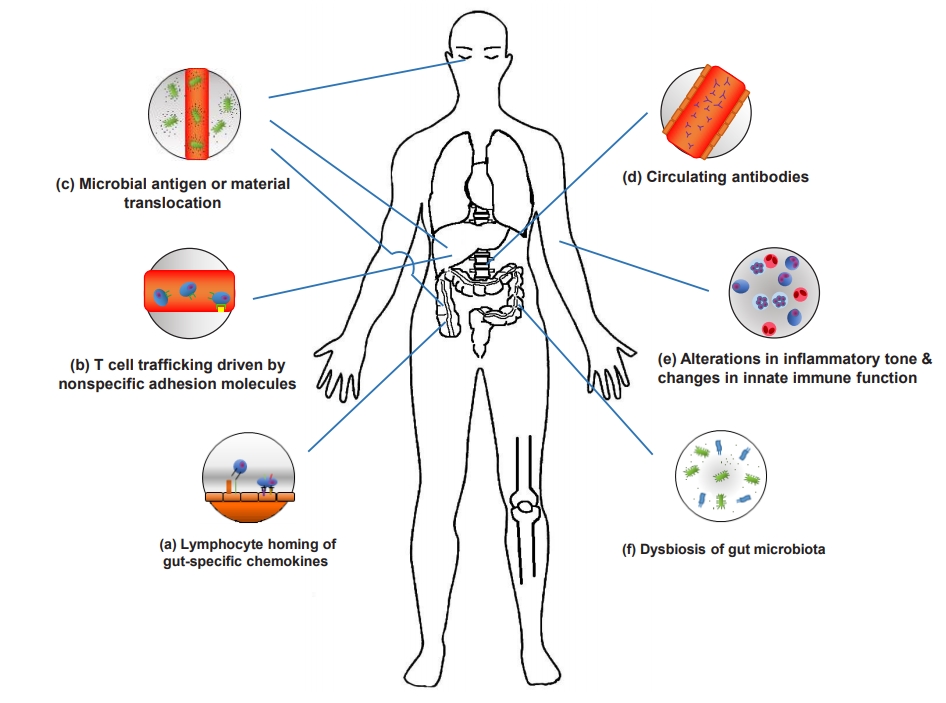
The pathogenic mechanisms of EIMs have yet to be clearly defined. Defining the pathophysiology of an EIM requires significant research and consensus due to the lack of consistent criteria for diagnosing an EIM and difficulty of distinguishing an EIM from extraintestinal complications or a secondary EIM caused by drugs. EIMs are considered to be the result of an antigen-specific immune response from the intestine to an extraintestinal site or an independent inflammatory event that is initiated or persisting from covalent genetic or environmental risk factors in the host, or from IBD (Fig. 1).
1. Dysbiosis of Gut Microbiota and Disrupted Gut Barrier in Intestinal Sites
Short chain fatty acids (SCFAs) enhance intestinal health, creating conditions favorable for resistance to pathogenic bacteria and preventing colitis. SCFAs have been found at significantly lower levels in IBD patients with psoriatic arthritis (PsA) [8]. IBD patients with PsA had a relative decrease in the abundance of Coprococcus and Ruminococcus species. Ruminococcus is a mucin-degrading bacterium that is especially important for maintaining intestinal homeostasis through SCFA production. The receptor activator of nuclear factor kappa-β ligand (RANKL)/ osteoprotegerin (OPG) system may be a regulator of intestinal microbiomes, and imbalances through the breakage of RANKL in the gut lumen of patients with PsA can promote dissociation of inflammation to a distal site, such as a joint [8].
IBD patients with spondyloarthritis (SpA) have an increased abundance of the genus Dialister, which is positively correlated with disease activity [9]. In addition, microbial activation in CD with SpA are associated with interleukin-23 (IL-23) dependent inflammation [10]. CD-SpA-derived Escherichia coli induced T helper 17 (Th17) cells polarization and made exacerbation of colitis or inflammatory arthritis.
Gut dysbiosis in IBD patients with AS induces differentiation and activation of group 3 congenital lymphocytes (IL-C3s) [11]. IL-C3s are IL-23-responsive immune cells that play a pro-inflammatory role in mediating a protective response against extracellular bacterial infections. IL-C3s have been identified in the peripheral blood, synovial fluid, and bone marrow tissues of patients with AS, most of which express intestinal homing integrin α4β7. Mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) was highly represented in the bone marrow and AS patients’ intestinal high endothelial venules. In addition, MAdCAM-1 plays a central role in attracting IL-C3s at the inflammation site of active AS and recirculating IL-C3s between the gut and inflamed spines [12]. In patients with IBD, intestinal effector T cells target gut microbial antigens. Transporting these antigens to the liver through the portal circulation allows the recruitment of mucosal effector α4β7 positive T cells through α4β7 MAdCAM-1 interactions.
Patients with PSC showed gut microbial differentiations from those with UC without liver diseases [13]. In a Gnotobiotic mice model with PSC/UC, Klebsiella pneumoniae, Proteus mirabilis and Enterococcus gallinarum were identified from the mesenteric lymph nodes of PSC/UC Gnotobiotic mice [14]. Disrupted gut barrier by K. pneumonia is associated with bacterial translocation and Th17 priming to the liver. These bacteria can lead the progression of hepatobiliary tract disease through the Th17 response in the colon and liver.
Alterations in the composition and metabolic products of the microbiota can affect the production of innate immune cells in patients with IBD [15,16]. There were significant differences in the composition of microbiome between HLA-B27+ and HLA-B27- siblings as well as healthy controls, indicating that genetic background can affect the composition and abundance of microbiomes.
Faecalibacterium prausnitzii-specific cells (DP8α TREG clones), found only in the chemokine receptor 6 (CCR6)+ and in the C-X-C motif chemokine receptor 6 (CXCR6)+ fractions of CD4CD8α (DP8α) cells, were significantly decreased in the blood of patients with IBD [17]. These gut recirculating markers are expressed in the DP8 cells of the large intestine and remain expressed in F. prausnitzii-reactive DP8α cells in the blood, allowing them to move to a specific intestinal position to perform their functions. These characteristics can be conveyed by the joint expression of CCR6 and CXCR6; the expression of CCR6 and CXCR6 also implies homing to other mucosal locations.
2. Immune Responses in Extraintestinal Sites
Abnormal lymphocyte homing in IBD can explain why EIMs are associated with site specific distributions [18]. The α4β7 integrin is a lymphocyte receptor for the MAdCAM-1. The interaction between α4β7 and MAdCAM-1 plays an important role in regulating lymphocyte homing to mucosal sites [19]. Expression of MAdCAM-1 is confined to the intestinal tissue, allowing gut tropic T cells expressing α4β7 integrin to selectively traffic with the intestinal mucosa. Additional tropism for the small bowel comes from C-C motif chemokine ligand 25 (CCL25) which attracts lymphocytes expressing the CCR9 receptor. T cell migration to the small bowel requires integrin α4β7 [19] and CCR9 [20]. Interestingly, homing to the colon requires α4β7 or α4β1 but not CCR9 [21].
Both chemokines and adhesion molecules can promote T cell trafficking to extraintestinal sites. The activation and expansion of effector memory T cells can lead to the induction of MAdCAM-1 and CCL25 expression in the liver, thereby promoting the recruitment of CCR9+α4β7+ mucosal T cells and development of inflammation [22]. Upregulation of inflammation-related adhesion molecules and chemokines also enables the capture of effector T cells, facilitating recruitment to sites other than extraintestinal sites. IL-6 produced during inflammation promotes the influx of naïve and memory T cells by increasing the expression of intercellular adhesion molecule-1 (ICAM-1) in high endothelial venules of the reactive lymph nodes [23]. ICAM-1 can also be upregulated with pre-inflammatory stimuli in response to TNF-α, IL-1β, and interferon-γ [24].
Neutrophils altered by inflammatory cytokines can enhance their response upon subsequent activation, referred to as “neutrophil priming [25].” Neutrophils in patients with gut inflammation are primed to secrete high levels of TNF-α and IL-1β after stimulation with lipid polysaccharides [26]. However, patients with CD have a defective neutrophil recruitment after the introduction of subcutaneous bacteria, which severely delays the subsequent clearance of these bacteria [27]. In addition to changes in circulating monocytes and macrophages in patients with IBD, reduced inflammatory cytokine production in response to bacterial stimuli has also been reported [27,28]. Neutrophil-platelet interactions play an important role in the degree of tissue neutrophil infiltration through neutrophil integrins (α1Lβ2 and αMβ2) and P-selectin glycoprotein ligand 1; however, neutrophil priming and/or activation in the peripheral site are inhibited by sphingosine [29,30]. Ceramide synthase 2 (CerS2) and CerS6 mutually control the aspects of neutrophil recruitment to tissues, and chemotaxis is also regulated by sphingosine-1-phosphate through IL-8 production. In the gut of patients with SpA, IL-23 polarization does not appear to be accompanied by Th17 polarization. However, IL-23 can induce gut inflammation and may be associated with the expansion and activation of innate immune cells such as IL-C3, γδ T cells, and mucosal-associated invariant T cells [11,31,32]. Augmented expression of IL-23R in γ/δ T cells was also associated with enhanced IL-17 secretion. Stimulation of γ/δ T cells from SpA patients with IL-23 and/or anti-CD3/CD28 strongly induced polarization for IL-17 production.
3. Autoantibodies
On the endothelial side, pre-incubation of the synovial sections with monoclonal antibodies against ICAM-1 and vascular adhesion protein-1 (VAP-1) showed significant inhibition of the binding of small lymphocytes from the IBD gut to the joint vessels [33]. Small lamina propria lymphocytes isolated from the intestine of IBD patients adhered to the synovial vessels using VAP-1 and their endothelial ligands (CD18 & ICAM1, and α4β7/α4β1 integrins and vascular cell adhesion molecule 1 [VCAM-1]). VAP-1 also plays a pivotal role in the recruitment and transmigration of lymphocytes via the hepatic sinusoidal endothelium, the expression of which is upregulated by inflammation [34]. VCAM-1 selectively binds lymphocytes through integrin α4β1. It also mediates lymphocyte homing to extraintestinal sites such as the lung, skin, and central nervous systems with chronic inflammation. It is not usually expressed by the intestinal venules but can be upregulated in the blood vessels during inflammation [35]. CCR10 with integrin α4β1 and VCAM-1 also mediate the localization of integrin α4β7-negative plasmablasts to non-intestinal mucosal sites (salivary and lacrimal glands and respiratory and urogenital tracts) where CCL28 is also expressed. Among the effector and central memory T cells, CCR10 is limited to integrin α4β1+α4β1- cutaneous lymphocyte antigen+ skin homing cells (which provide T cell attraction to the CCR10 ligand CCL27 expressed in keratinocytes). Inflammation or mechanical stress in the extraintestinal sites can lead to the recruitment of gut-producing effector T cells and further increase the inflammatory process [34,36].
In addition, molecular mimicry, peptide sequences between the gut bacteria and host major histocompatibility complex (MHC) molecules, and reactivation by cross-reactive components of microbiomes or host antigens have been reported [37,38].
Peptides expressing common epitopes shared by colons and extra-colonic organs, autoimmune responses to the isoform of tropomyosin (tropomyosin related peptide) expressed in the eyes, skin, joints, biliary epithelia, and intestines can help clarify the pathogenesis of EIMs [39,40]. Bacteria can translocate across the leaky epithelial barriers and cause adaptive immune responses that are indistinguishable from bacterial epitopes in joints or skin. There is still a lack of clear evidence on the role of circulating antibodies or immune complexes in the pathogenesis of EIMs.
In a mice model, retina-specific T cells causing human blindness due to uveitis received microbial-dependent activation signals, most likely cross-reactive bacterial antigens [41]. This result showed that the activation of autoreactive T cells via gut microbes may drive uveitis. In another mice model of autoimmune uveitis, enhanced leukocytes trafficking between the intestine and eyes were observed [42]. The antigen specificity of T cells responsible for EIMs in humans will require further confirmation.
4. Genetics of EIMs
Genetic risk loci for both IBD and EIMs overlap extensively. PSC genes and their linked IBD genes are involved in T cell apoptosis (UBASH3A, BCL2L11, FOXO1, and IRF8) and the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways signaling pathways (SOCS1, JAK2, STAT3, and TYK2). In case of AS, T cell apoptosis pathways interact with the IBD genes. The AS-related genes, TAPBPL and NPEPPS, function similarly to the AS covalent gene ERAP1 in patients with IBD, which is important for both the development of AS and IBD. ERAP1 variant K528R associated with CD has involved in aberrant peptide production resulting in suboptimal peptide MHC I complex [43]. The CD risk gene, NOD2/CARD15, which encodes a pattern recognition receptor, is also associated with sacroiliitis and uveitis [44,45]. STAT4 is also responsible for the expansion of Th17 cells activated by IL-23, which promotes chronic inflammation in innate and adaptive immunity and is considerably associated with the development of EIMs involving the joints and eyes in patients with IBD [46]. However, it is still unknown whether all relevant loci contribute specifically to the pathology of EIMs or whether there are genes that generally cause EIMs by not restricting the inflammatory response to specific body compartments. Therefore, further research is needed.
CLINICAL ASPECTS OF EIMs
This following section describes the paradigms and tools currently available in clinical studies of the musculoskeletal/joint, skin, eye, vessels, and hepatobiliary EIMs (Table 2).
1. Musculoskeletal/Joint Manifestations
Inflammatory arthrosis of IBD is the most common EIM, with a prevalence of 20%-50% for axial inflammation and 5%-20% for peripheral arthritis [47]. Peripheral arthritis in patients with IBD is mainly asymmetric, oligo-articular, and is predominantly found in CD. IBD-related joint symptoms can be subdivided into non-inflammatory and inflammatory joint pain. Arthralgia is a major medical problem in patients with IBD. Compared to patients with IBD without arthralgia, non-inflammatory joint pain was reported in 16% of patients with a significant worsening of their HRQOL determined using the 36-Item Short-Form Health Survey and the Inflammatory Bowel Disease Questionnaire [48]. The current gold standard tool for layering SpA is the Ankylosing Spondylitis Disease Activity Score, which can measure the disease activity that may be followed over time [49]. In general, the prognosis for peripheral arthritis is good. The prognosis for axial involvement is less favorable and is not related to the prognosis of IBD but to the progression of IBD.
2. Skin Manifestations
Diagnoses of skin manifestations are primarily based on clinical examinations, and skin biopsies can help in atypical cases. EN is associated with IBD activity, arthralgia, or fatigue. The prevalence of EN appears higher in patients with CD than in those with UC [50]. EN usually appears on the extensor surface of the lower extremities, mostly in the anterior tibial region [51], and is associated with IBD flares but not always with severity, is self-limited, and usually resolves itself without the formation of ulcers or scarring [52]. The appearance of nodules parallels intestinal activity, which rarely occurs before the onset of IBD or in the quiescent phase of IBD. IBD treatment leads to the resolution of cutaneous lesions. PG can occur anywhere in the body but occurs most commonly in the shin and adjacent to stomas. PG usually has a skin trauma known as “pathergy.” This trauma might even be minimal, such as a venipuncture. Controversy exists over whether PG coexists with disease activity. In this case, treatment can be concentrated on the underlying intestinal disease. Patients often show a rapid response, with a stiffening of the lesions within 24 hours of treatment.
Due to the absence of specific tests for diagnosing PG, physicians can determine whether clinical and histopathological images are consistent with PG, excluding other pustules or ulcerative dermatitis. A further development of assessment tools and standardized classifications are needed.
3. Eye Manifestations
The most common ocular EIMs are dry eye, blepharitis, episcleritis, or anterior uveitis. Some ocular manifestations in IBD can be secondary to treatment and/or effects of the IBD itself. Ophthalmic complications usually originate from inflammation, but some of these complications may reflect the overall disease activity [53]. Episcleritis can be unilateral or bilateral and is more common in women. It usually does not need specific treatments other than those for the original disease. Scleritis is chronic and is characterized by edema and cellular infiltration of the scleral tissues. Uveitis is not common but can have potentially more serious consequences. It occurs acutely or subacutely and is usually very painful. Anterior uveitis is also commonly referred to as iritis, which indicates pain and photophobia and can be associated with blurred vision or floaters. The slit-lamp test confirms the diagnosis of an ocular EIM. In case of vision impairment, the patient must be immediately referred to the ophthalmologist to avoid vision loss.
4. Vascular Manifestations
Cardiovascular manifestations in IBD mostly occur as an immune-related pathogenesis including pericarditis, myocarditis, venous and arterial thromboembolisms, arrhythmias and conduction disorders, infective endocarditis, and Takayasu’s arteritis. Pericarditis is the most frequent cardiovascular EIM in patients with IBD but has not been associated with IBD activity [54]. The clinical picture of myocarditis or pericarditis related to IBD is nonspecific. These patients may have symptoms of an acute coronary syndrome, heart failure, or arrhythmias and may also have cardiogenic shock or sudden death.
If this clinical picture occurs within the first 28 days after starting treatment with 5-aminosalicylic acid or its derivatives, drug toxicity is suspected [55]. Because 12-lead electrocardiograms and biomarkers of cardiac injury are also nonspecific, transthoracic echocardiography, coronary angiography, or cardiovascular magnetic resonance imaging may offer more precise information for all patients with clinical presentations suggesting myopericarditis [56]. Furthermore, endomyocardial biopsies can be considered a gold standard for the diagnosis of myocarditis [57].
In meta-analysis studies, the risk of arterial thromboembolic events or cardiovascular mortality in patients with IBD does not increase but is associated with an increase in the risk of cardiovascular morbidities from cerebrovascular accidents and ischemic heart disease, especially in women [58,59]. Lower rates of peripheral vascular disease in IBD do not appear to increase the risk of peripheral arterial thromboembolic diseases. However, hyperhomocysteinemia, a risk factor for arterial and venous thrombosis, is more common in patients with IBD than in the general population [60]. In addition, an increased risk of arterial thrombosis due to systemic inflammation and hypercoagulability involves premature atherosclerosis [61].
5. Hepatobiliary Manifestations
Approximately 20% to 30% of patients with IBD show abnormal hepatic biochemistries [62]. PSC is highly correlated with IBD, and about 50%-80% of patients with PSC have an accompanying IBD [63]. Patients with IBD and PSC often have predominantly right-sided, high-frequency “backwash” ileitis, and rectal sparing [64]. Patients with UC, when diagnosed with PSC, are negatively associated with smoking and older ages [65]. Symptoms are intermittent and can be confusing due to the underlying IBD symptoms. In patients with IBD, PSC is usually asymptomatic, which requires an awareness of high clinical association. Although typically imaged using magnetic resonance cholangiography and confirmed using endoscopic retrograde cholangiopancreatography (ERCP), the Crohn’s and Colitis Organisation consensus group suggests that ERCP be performed only for intervention in stricture dilatation and/or if brush cytology specimen sampling is indicated [66].
Autoimmune hepatitis (AIH)/PSC overlapping syndrome is seen in UC patients than CD [67]. It is suspected that the characteristics of AIH and PSC in the same patient are present, and a definitive diagnosis of AIH is required according to the international AIH group criteria [68].
The prevalence of AIH in pediatric IBD patients is 1.6% [69]. AIH can be divided into 2 types. AIH type 1 is positive for antinuclear antibodies and/or smooth muscle antibodies, whereas AIH type 2 is liver/kidney microsomal type 1 and/or antiliver cytosol type 1 is positive. Even if antinuclear antibodies or smooth muscle antibodies is 1:20 or liver/kidney microsomal type 1 is 1:10 in children, it is associated with AIH in children [70].
Granulomatous hepatitis is a rare EIM of CD. The estimated prevalence is less than 1% of IBD. It may manifest fever, alkaline phosphatase, hepatosplenomegaly. Granulomatous hepatitis may be induced by mesalazine and sulfasalazine therapy [71].
In nonalcoholic fatty liver disease, IBD-specific risk factors include intraperitoneal abscesses, fistula disease, colitis severity, malnutrition, protein loss, and drug use.
Thiopurine and gallstones are the most common causes of acute pancreatitis in patients with IBD, and acute pancreatitis caused by thiopurine is generally not complicated and is selfl-imiting [72]. Most chronic pancreatitis cases in patients with IBD are idiopathic, and PSC is a less common cause. Exocrine pancreatic insufficiency appears to be the most common pancreatic symptom in IBD, but its clinical significance remains unknown.
TREATMENT FOR EIMs INCLUDING BIOLOGICS
Optimizing treatments is often challenging. There is lack of high-quality evidence to support the most commonly used treatment methods for EIMs and other treatment strategies that can complement them. Treatment of EIMs should be based on the severity of symptoms and their association with IBD activity. The primary goal is symptom control and preservation of mobility and function. In some cases, symptomatic treatment with physiotherapy may be sufficient (Table 3).
1. Joint Manifestations
Treatment of IBD-related arthropathy is based on SpA studies, predominantly AS. Patients with axial SpA should be co-managed by rheumatologists because of the potentially disruptive disease progress. Severity of type 1 arthritis usually runs parallel to IBD activity, and type 2 arthritis usually requires long-term treatment. Therefore, effective treatment of underlying gut inflammation is important for the treatment of peripheral arthritis.
Intensive physiotherapy may be helpful in AS, and NSAIDs should be used for a short time period due to their potential to activate underlying IBD [73]. Cyclooxygenase-2 inhibitors such as etoricoxib and celecoxib may be safer and have a lower risk of disease flare than conventional NSAIDs [74]. Sulfasalazine, methotrexate (MTX), and azathioprine are generally ineffective or only slightly effective in the treatment of AS. However, sulfasalazine is effective in treating patients with peripheral joint disease, especially those with a short disease duration [75]. Sulfasalazine is an optional treatment in patients with SpA and peripheral disease and may be effective for large joint arthropathy [76]. MTX and azathioprine are considered marginally effective in the treatment of peripheral arthropathy. For patients with intolerance, unresponsiveness, or poor response to NSAIDs, anti-TNF therapy is the preferred treatment because of the limited efficacy of MTX and thiopurine [77].
2. Skin Manifestations
Treatment of EN consists of treating the underlying IBD, and systemic steroids may be helpful. For patients with resistant or frequent relapses, immunomodulators and anti-TNF therapy can be considered alone or in combination [78].
PG treatment strategies are the same between IBD and non-IBD patients. Systemic corticosteroids are considered as the first-line treatment, and oral or intravenous tacrolimus with oral cyclosporines may be considered for refractory cases [79,80]. Anti-TNF therapy represents a new and effective treatment option for medically refractory PG cases.
3. Eye Manifestations
Due to the potential loss of vision, ophthalmologists must evaluate each suspected case to confirm the diagnosis and help guide the treatment. Dry eyes may be treated with topical lubricants. Episcleritis may be self-limiting and usually responds to underlying IBD treatment along with topical NSAIDs and glucocorticoids [81]. Uveitis requires urgent consultations with ophthalmologists and needs prompt treatment because of the risk of vision loss. Treatment usually consists of steroids. Other therapies (azathioprine, MTX, or anti-TNF therapy) have been discussed only for resistant cases [82].
4. Vascular Manifestations
Because thromboembolism occurs due to multifactorial causes and acquired risk factors, precautions (hydration, correction of vitamin deficiencies, and wearing gradual compression stockings) can help prevent venous thromboembolisms (VTEs). Prophylactic anticoagulation therapy is recommended for high-risk patients (hospitalized due to an active phase or surgery) with VTEs [83]. There is still no evidence for the prophylaxis of active disease in outpatients with IBD. However, because many VTEs occur in outpatients with IBD, guidelines are required for VTE prophylaxis when patients with IBD have acutely or chronically active diseases. Acute deep vein thromboses and pulmonary embolisms are treated using anticoagulants according to international guidelines. Low-molecular-weight heparin is commonly used, and low-dose unfractionated heparin or fondaparinux can be used as an alternative option. Long-term treatment includes oral anticoagulants such as vitamin K antagonists (acenocoumarol, warfarin, fluindione) or non-vitamin K antagonist oral anticoagulants, such as dabigatran, rivaroxaban, apixaban, or edoxaban.
5. Hepatobiliary Manifestations
Ursodeoxycholic acid improves abnormal liver function tests and is widely used in patients with PSC, but prognosis improvements are controversial. Other therapeutic agents (steroid, cyclosporine, tacrolimus, MTX, or anti-TNF therapy) still lack evidence for improvements in clinical endpoints [84]. ERCPs play a role in the management of biliary strictures and should be performed after antibiotic prophylaxis in patients with PSC. It can reduce the risk of cholangitis, bacteremia, and septicemia after ERCP [85]. Patients with PSC and end-stage liver disease should be evaluated to determine whether liver transplantations should be performed according to the standard guidelines. PSC has a significantly greater risk for the development of colorectal dysplasia and carcinoma in patients with UC [86]. Therefore, surveillance colonoscopies with target biopsies are recommended at the diagnosis and at every 1-2 years thereafter [87].
Immunosuppressive therapy with prednisolone alone or in combination with azathioprine is the main therapeutic regimen of AIH and has been to show the prolongation of life expectancy [67]. In granulomatous hepatitis, corticosteroids and immunosuppressive drugs are used for treatment.
6. Biologics and Probiotics for EIMs
Biologics have been tested to prove their efficacy in the treatment of EIMs as well as IBD. Clinical benefits of infliximab or adalimumab may not be limited to gut treatment effects but may extend to improve EIMs [88]. Infliximab and adalimumab therapy also have potential beneficial effects on metabolic bone diseases, hematologic diseases, or vascular EIMs [88]. However, anti-TNF treatments can cause paradoxical cutaneous inflammations, which are considered a class drug effect, and are generally reversible upon drug transition or discontinuation. This paradoxical cutaneous inflammation manifests itself or appears in most cases as psoriasis, which accounts for why anti-TNF-α drug use is increasingly recognized as a cause of drug-induced skin lesions [89]. Paradoxical psoriasis skin lesions are characterized by selective overexpression of type I interferon, skin accumulation of plasmacytoid dendritic cells (pDCs), and reduced T cell counts [90]. Anti-TNF-α treatment prolongs the production of type 1 interferons by pDC by inhibiting pDC maturation. Type I interferon overexpression results in a skin phenotype of paradoxical psoriasis, independent of T cells, unlike classical psoriasis. TNF-α inhibition may reduce the accumulation of Th1 and Th17 cells at the site of inflammation but may induce compensatory expansion at other locations, resulting in skin lesions [91].
The role of vedolizumab in EIM treatment has not been defined yet. Vedolizumab inhibits the binding of α4β7+ integrins to the intestinal mucosal cell adhesion molecule (MAdCAM-1) but not to VCAM-1 [92]. The binding mechanism seems to be selective for cutaneous intestinal lymphocyte migration. In contrast, lymphocyte migration by VCAM-1 is unaffected. Nevertheless, the overlapping binding behavior of α4β7 with MAdCAM-1 and VCAM-1 may be due to partly different affinities [92,93]. According to these results, vedolizumab has no direct therapeutic effect on cutaneous EIMs and can play a role in EIM management by treating disease activity in EIMs that parallels IBD [94]. In a systematic review, vedolizumab was suggested to reduce the incidence of new EIMs but also be effective in the treatment of preexisting EIMs (particularly PSC, rheumatic and cutaneous manifestations) [95].
Data on other biologicals are limited. Ustekinumab is a fully human IgG1 kappa monoclonal antibody that blocks the IL-12/23 p40 subunit. IL-12 and IL-23 are associated with adaptive immune responses, and blocking IL-12/IL-23 helps improve chronic inflammatory conditions. Ustekinumab has been reported for the management of cutaneous and joint EIMs (psoriasis, PsA) [96,97]. In addition, it is effective for the treatment of anti-TNF antibody-related paradoxical cutaneous inflammation [98]. However, paradoxical arthralgic or cutaneous manifestations have also been reported [99,100]. In addition, it did not reduce the occurrence of new EIMs [101]. Therefore, ustekinumab requires additional safety and efficacy evidence from randomized controlled trials and open-label, real-world cohorts. Ustekinumab will be an important treatment option in EIMs and IBD because it is superior to vedolizumab in terms of overall efficacy, rapid onset, and efficacy in small intestine disease.
Tofacitinib is a selective JAK inhibitor and the first oral medication approved for use in treating moderate-to-severe UC. Its efficacy of treating cutaneous EIMs has been reported. STAT3 was significantly upregulated in PG/EN compared to psoriasis and revealed potential benefits from JAK inhibition in these patients [94,102]. In a double-blind, head-to-head, non-inferiority, randomized controlled trial in patients with rheumatoid arthritis who had insufficient response to MTX treatment, combination therapy with tofacitinib and MTX showed similar efficacy to that with adalimumab and MTX, and tofacitinib monotherapy was shown to be inferior to either combination [103]. Because tofacitinib is expected to be free of anti-drug antibodies due to its low molecular weight, it results in a rapid therapeutic effect in patients with acute severe UC. Further studies on the efficacy and safety of the combination therapy with tofacitinib and other drugs are needed. In addition, there is a need to compare the efficacy and side-effects of upadacitinib and tofacitinib, which are selective JAK1 inhibitors.
KEY RECOMMENDATIONS FOR IMPROVING THE DISEASE COURSE AND PRINCIPLES TO BE APPLIED TO TREATMENT
Physicians should always consider EIMs in the management of patients with IBD. Early diagnosis and treatment can prevent permanent tissue destruction and the development of complications in patients with IBD with developing EIMs. Diagnostic delay is considered a risk factor for the development of EIM complications and abdominal surgery [106]. Uveitis can result in visual impairment if the diagnosis is delayed. Due to the diagnostic delay accompanying these inflammatory conditions, the need for proper approaches and tools that can quickly provide accurate and viable information is further emphasized. Sex, age, ethnicity, geography, socioeconomic status, duration, and extension of disease, disease activity, and a history of medications are associated with the diagnoses of EIMs. Genetic factors and dysbiosis of gut microbiomes also play an important role in identifying EIMs. Physicians should be careful because specific features of the clinical presentations may signal the possibility of future EIMs. Screening and surveillance of EIMs are difficult for physicians in only one specialty to include in their treatment protocols. Therefore, it is necessary to follow-up with multidisciplinary care. Educating patients on EIMs and their complications will improve the effectiveness of medications and follow-ups. However, research has shown that certain EIMs, such as VTEs, PSCs, and nephrolithiasis, may be less emphasized in patient education [107]. Patients’ perception of thromboembolisms or cardiovascular EIMs can have a significant impact on the associated morbidity and mortality. VTE has a higher incidence in patients with IBD than in the general population, and IBD disease activity is associated with an increased risk of VTE [108]. However, only 12% of patients were aware that the risk of VTE in the outpatient environment was high and less than half of the patients were convinced that they recognized the signs and symptoms of deep vein thromboses and pulmonary embolisms [107], indicating a need to reconsider the awareness of EIMs in patients with IBD.
The role of mobile applicable for the self-management of IBD by patients will continue to increase by providing a more efficient, effective, and good quality healthcare system at lower costs [109]. Daily monitoring of UC through TrueColours UC improved UC management while also reducing the burden placed on individual and healthcare services [110]. These applications can not only remotely monitor patients via electronic patient-reported outcome measures (PROs) or device-generated data (such as activity trackers) but also improves patient engagement [111]. The goal of recent IBD therapies is moving toward a therapeutic-targeted strategy using PROs, biomarkers of inflammation, and mucosal healing to optimize therapeutics and should also be used for the treatment of EIMs.
CONCLUSIONS
For EIMs of IBD, new mechanisms are being uncovered in a number of fields, including immunology, microbiology, and genetics, and can reveal a wide range of therapeutic targets. It is fundamentally important to ensure proper diagnosis and management of EIMs, which can often result in a much more debilitating and lower quality of life than actual IBD. Better patient images require new tools to monitor inflammation from various angles, and managements for complex and diverse EIMs should be discussed, treated, and tracked through multi-disciplinary care. In addition to establishing the required diagnosis and treatment process, patients should be encouraged to participate more actively in the various stages of medical care, measurements, implementations, and evaluations of the quality of their care.