Intestinal microbiota, chronic inflammation, and colorectal cancer
Article information
Abstract
In addition to genetic and epigenetic factors, various environmental factors, including diet, play important roles in the development of colorectal cancer (CRC). Recently, there is increasing interest in the intestinal microbiota as an environmental risk factor for CRC, because diet also influences the composition of the intestinal microbiota. The human intestinal microbiota comprises about 100 trillion microbes. This microbiome thrives on undigested dietary residues in the intestinal lumen and produces various metabolites. It is well known that the dietary risk factors for CRC are mediated by dysbiosis of the intestinal microbiota and their metabolites. In this review, we describe the bacterial taxa associated with CRC, including Fusobacterium nucleatum, enterotoxigenic Bacteroides fragilis, Escherichia coli, and butyrate-producing bacteria. We also discuss the host-diet interaction in colorectal carcinogenesis.
INTRODUCTION
Colorectal cancer (CRC) is the second most common cancer in women and the third most common cancer in men worldwide.1 CRC is regarded as a Westernized disease, as it has the highest incidence rates in North America, Australia, New Zealand, and Europe.2 However, over the last several decades, the incidence rates of CRC in the United States have been decreasing.3 This reduction is thought to be due to changes in the prevalence of risk factors and increased screening.4 In contrast, in Asian countries, including Korea, the incidence of CRC has been increasing in both men and women.56
CRC can develop via various pathways. More than 70% of CRC cases develop from premalignant adenomas in a multistep genetic process. Some forms of CRC are hereditary, but these represent less than 5% of total cases. In addition to genetic susceptibility, epigenetic mechanisms are also involved in colorectal carcinogenesis. For example, the epigenome of CRC usually has hundreds to thousands of abnormally methylated genes, and a subset of these are thought to initiate the development of CRC.7 In additional to genetic and epigenetic factors, the intestinal microbiota is increasingly recognized as a confounding factor in the development of CRC. Finally, other environmental factors, such as diet, also play an important role in the development of CRC.8 For example, high dietary fiber intake is associated with a reduced risk of CRC, while high intake of red meat and processed meat is associated with an increased risk of CRC.910 The International Agency for Research on Cancer (IARC), the cancer agency of the World Health Organization (WHO), reported the risk of excessive consumption of red meat and processed meat.11 A study on colon cancer risk in Japanese immigrants showed progressive changes in the incidence of colon cancer among Japanese after migrating to Hawaii.12 Such evidence supports a strong relationship between diet and the development of CRC.
Recently, interest in the intestinal microbiota as an environmental factor for CRC has increased, because diet also influences the composition of the intestinal microbiota. The human intestinal microbiota is a community of approximately 100 trillion microbes13 that thrives on the undigested dietary residues in the intestinal lumen and produces various metabolites. It is well known that the dietary risk of CRC is mediated by dysbiosis of the intestinal microbiota and their metabolites.14 The relationship between diet and intestinal bacteria was also demonstrated in a metagenome-wide association study. High intake of red meat relative to fruits and vegetables was shown to be associated with the outgrowth of bacteria that may contribute to a more hostile gut environment.15 In addition, it has been shown that there are similar bacterial networks on oral and colonic mucosal surfaces, in both individuals with colonic lesions (on and off the tumor) and healthy controls.16 These findings support the notion that colonic bacteria may be derived from dietary food and oral bacteria.
BACTERIAL TAXA ASSOCIATED WITH CRC
There are few data showing that the intestinal microbiota directly induce CRC. However, it has been suggested that some bacteria have a pivotal role of the development of CRC. It has been shown that Fusobacterium nucleatum is associated with intestinal tumorigenesis and modulates the tumor-immune microenvironment.17 Many case-control studies have demonstrated that the abundance of F. nucleatum was higher in patients with CRC than in controls.18192021 In addition, several studies have reported that F. nucleatum is associated with colorectal adenoma rather than CRC.2223 Patients with CRC harboring F. nucleatum in their mucosa have more advanced disease and poorer prognosis, which is associated with the response to chemotherapeutic agents. F. nucleatum can affect host immunity through its attachment to epithelial cells via Fusobacterium adhesion A (FadA),24 and its recognition of the Gal-GalNAc portion of the membrane by the lectin Fap2.25 These interactions lead to bacterial adhesion and invasion, as well as an altered immune response to bacterial infection, via decreased apoptosis, cellular proliferation, and DNA repair, through activation of the nuclear factor-κB(NF-κB) and Wnt signaling pathways. Increased expression of LC3-II mediated by F. nucleatum induces the expression of autophagy-related proteins.26 This affects host immune surveillance and orchestrates Toll-like receptors, microRNAs, and the autophagy network to control cancer chemo-resistance. As a result, in CRC patients that harbor F. nucleatum, the cancer frequently recurs after drug therapy.26 In addition, F. nucleatum produces a protein that binds to an immunoreceptor on T and natural killer cells and blocks their cytotoxic activity against tumor cells.27
Fecal transfer from mice with CRC promotes colon carcinogenesis. In a recent study of fecal transfer from mice with CRC to healthy mice, F. nucleatum and Peptostreptococcus anaerobius were relatively abundant.28 After fecal transfer, increases in the number of polyps and the levels of cellular dysplasia, epithelial proliferation, inflammatory markers, and Th1 and Th17 cells were noted. These findings indicate that the intestinal microbiota has a strong influence on host immunity via cross-talk. Microbes like F. nucleatum may affect the development of CRC through the metabolites they produce, such as the short chain fatty acid (SCFA). SCFAs, principally butyrate, propionate, and acetate, have a remarkable array of colonic health-promoting and antineoplastic effects.2930 Although the detailed mechanism of F. nucleatum-driven CRC development via butyrate metabolism is not well understood, a metagenome-wide association study detected butyryl-CoA dehydrogenase from F. nucleatum as a genetic marker for CRC.31 Colorectal carcinogenesis is accompanied by rupture and bleeding of the cancerous tissue, which alters the microenvironment and puts selective pressure on the local microorganisms.32 These changes facilitate the gradual replacement of the typical commensal intestinal microorganisms by F. nucleatum.
In addition to F. nucleatum, a variety of other bacteria are associated with CRC in mice. For example, enterotoxigenic Bacteroides fragilis (ETBF), polyketide synthase (pks) NC101 Escherichia coli, Streptococcus gallolyticus, and Enterococcus faecalis have been shown to promote the development of CRC in in vitro studies. Bacteroides is an abundant microorganism in the intestine of patients with CRC. ETBF encodes a specific virulence factor, the 21 kDa B. fragilis toxin, that cleaves E-cadherin on the host epithelium, affects epithelial permeability, and causes intestinal inflammation.33 In a pyrosequencing analysis, although the absolute abundance of bacteria per gram of stool in CRC patients and healthy individuals was similar, the abundance of Bacteroides was higher in patients with CRC than in healthy individuals.34 In addition, an operational taxonomic unit (OTU) closely related to B. fragilis was enriched in the gut microbiome of CRC patients.35 Another study showed that the abundance of Bacteroides was increased in colorectal adenoma.36 ETBF secretes an enterotoxin that functions as a zinc-dependent metalloprotease and causes DNA damage.37 In apc-deficient mice, this enterotoxin leads to interleukin 17 (IL-17)-dependent inflammation and induces the development of distal CRC. Additionally, infection with ETBF causes chronic colitis, with robust and rapid colonic activation of signal transducer and activator of transcription 3 (STAT3).3839 ETBF also increased the number of adenomas in early stage CRC in a mouse model. Based on these findings, several OTUs from Bacteroides have been implicated as microbial biomarkers for the prediction of colorectal neoplasia.36 In fact, the combination of metagenome analysis and the fecal occult blood test (FOBT) increased the sensitivity for the prediction of CRC by 45% compared to FOBT alone.19
Some strains of E. coli detected in CRC are more invasive than the strains observed in other diseases. For example, E. coli harboring pks islands, which are more common in CRC than are bacteria that do not have pks islands,40 produce colibactin, a genotoxic metabolite (encoded by pks island) that promotes SUMO-conjugation to p53 tumor suppressor protein and cell senescence.3341 Thereafter, it induces hepatocyte growth factor production and enhanced tumor cell proliferation.3341 In mouse enterocytes, infection with pks+ E. coli resulted in DNA damage.42 Mono-colonization of carcinogen-treated IL-10-deficient mice with E. coli harboring pks islands induced the development of invasive CRC.40 In addition, the pks island has been shown to be required for tumorigenesis in the gut.4043
Another species, S. gallolyticus, which was formerly known as Streptococcus bovis, is well-known to be involved in cases of bacterial endocarditis associated with CRC. Approximately 18% of patients who were infected with S. gallolyticus (most of whom had infective endocarditis) had CRC.44 When colorectal adenoma or cancer develops, distortion of the colonic mucosa may allow bacteria, such as S. gallolyticus, to access the previously unexposed collagen fibers in the basement membrane.45
As mentioned above, various bacterial taxa have been shown to be associated with CRC. These findings have been supported by a metagenome-wide association study.15 In the study, approximately 59% of the gene markers were significantly more abundant in CRC when compared with both controls and advanced adenomas. Another 24.3% of the genes were significantly elevated when compared with the controls. In contrast, only 4.1% of genes were significantly decreased in CRC when compared with the controls and advanced adenomas. This disparity in the number of increasing and decreasing genes suggests that the increase in pathobionts is more pronounced than the decrease in beneficial bacteria in colorectal carcinogenesis.15
Some bacterial taxa are associated with a lower risk of CRC. For instance, butyrate-producing bacteria, including Lachnospiraceae and Clostridium, are thought to be negatively associated with CRC. Through 16S rRNA gene sequencing analysis, Roseburia and other butyrate-producing bacteria of the family Lachnospiraceae and genus Clostridium were shown to be less abundant in patients with CRC.3536 These bacterial taxa are well known producers of SCFAs in the colon.36 SCFAs are the preferred energy substrate for colonic cells, and they help maintain epithelial health and homeostasis.3646 Additionally, butyrate has substantial anti-tumorigenesis effects, including inhibition of tumor cell proliferation, initiation of apoptosis in tumor cells, and mediation of T-regulatory cell homeostasis.4748 Loss of butyrate-producing bacteria and enrichment of pathogenic bacteria may have synergistic effects on tumorigenesis.36
The numbers of lactic acid-producing bacteria, such as Bifidobacterium animalis and Streptococcus thermophilus, are decreased in feces from patients with colorectal adenoma or cancer.15 The lactic acid produced by these bacteria may increase intraluminal acidity in the colon and inhibit amino acid degradation.4950 Lactic acid-producing bacteria, including Bifidobacterium, have been shown to stimulate the generation of nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) 1-dependent reactive oxygen species and intestinal stem cell proliferation.51 In addition, lactic acid accelerates turnover of the colonic epithelium in starvation-refed mice.52 In patients with a lower abundance of lactic acid-producing bacteria, this daily renewal of the colon epithelial cells may be prevented, allowing potential pathogens to grow.
Although many studies have demonstrated an association between certain bacteria and the development of CRC, not all studies have yielded consistent results.53 Several studies suggested Fusobacteria as an abundant bacterial taxon in patients with CRC, while other studies identified Bacteroides as the major bacterial taxon associated with CRC.53 In addition, it is unlikely that there is only one key player in the development of CRC. Therefore, there have been attempts to classify the bacteria that affect CRC development using clustering methods. Flemer et al.54 divided identified bacterial taxa into Pathogen cluster, Bacteroides cluster 1, Bacteroides cluster 2, Firmicutes cluster 1, Firmicutes cluster 2, and Prevotella cluster. In the study, the pathogen cluster consisted of Fusobacterium, Parvimonas, and Peptostreptococcus, while butyrate-producing bacteria, such as Clostridium, were included in the Firmicutes clusters.
METHANOGENIC ARCHAEA AND CRC
Methanogenic archaea is identified in about 25% to 40% of children and 42% to 82% of adults.5556 It the colon of healthy individuals, methanogenic archaea can constitute up to 10% of all anaerobes.57 However, it can also play a role of pathogen in the colorectal carcinogenesis. Although the mechanism is still doubt, it seemed to be associated with a microbial shift towards an anaerobic consortium, consisting of saccharolytic and proteolytic anaerobic bacteria, including Clostridium, Eubacterium, the Bacteroides/Prevotella cluster, the terminal-degrading methanogens, and sulfate-reducing bacteria, with a concomitant reduction in probiotic bacteria.58
MICROBIAL DISTRIBUTION AND ANATOMICAL DIFFERENCES
The intestinal epithelium is covered by mucus layer. The mucus in the large intestine is divided into a 2-layered structure composed mainly of the mucin MUC2.59 The inner mucus layer is very dense, free of bacteria, and plays a role in preventing the invasion of microbes into the epithelial cells. The outer mucus layer is very thick and is heavily colonized by resident bacteria.60 Mucin glycan plays a key role in controlling the microbial community.61 Bacterial mucin glycan catabolism is important for gut colonization, and unexpected changes in mucin-degrading bacteria may interrupt gut homeostasis.61 Intestinal mucus is closely related to diet, and in mice, a low-fiber diet accelerates degradation of mucus glycan and weakens this layer.62 Biofilms generated by resident microbiota can modify epithelial cell biology and are associated with the initiation and progression of CRC.63 Much greater biofilm growth is detected in colon cancer tissues, especially in proximal colon cancer tissues. Bacterial biofilm formation triggers prolonged activation of inflammation.64 Although various mechanisms regarding biofilm formation and CRC have been proposed and are currently unproven, evidence suggests that biofilm formation and its association with intestinal dysbiosis may be an initial process in colorectal carcinogenesis.65
Based on the overwhelming burden of microbial biofilms in the colon proximal to the hepatic flexure, Dejea et al.63 proposed redefining the proximal colon as the cecum, ascending colon, and hepatic flexure. In addition, the microbial load and levels of metabolites such as SCFAs also differ between proximal and distal CRCs.66 A study of 1,102 CRCs using quantitative PCR found that the proportion of F. nucleatum-high CRCs gradually increased according to location (2.5% for rectal cancers to 11% for cecal cancers), with a statistically significant linear trend along all subsites.67 These differences in microbial distribution and biofilms may lead to molecular differences in the development of CRCs between the proximal and distal colon.
HOST-DIET INTERACTION
Despite the known association between F. nucleatum and CRC, there is no evidence that F. nucleatum is involved in the early stage of colorectal carcinogenesis. Multiple bacteria that compete for luminal nutrients, rather than a single bacterium, may influence the development of CRC. Therefore, more studies are needed to identify the causative roles of the microbiota in colon carcinogenesis in humans.
Lifestyle and metabolic diseases are well-known risk factors for CRC. Diet affects the composition of the gut microbiota, and dysbiosis has been shown to be associated with various diseases, such as obesity, IBD, colon cancer, and so forth. Longstanding diet habits can change the microbiota and lead to defects in the inner mucus layer and compromised epithelial integrity. Such altered epithelial integrity allows translocation of bacteria and diffusion of microbiotaderived metabolites. These microbes ferment undigested intraluminal fibers as an energy source and produce SCFAs, which are essential for epithelial cell homeostasis. SCFAs also control microRNA expression and histone deacetylation. Changes in histone deacetylation can alter cell cycle regulation.68 SCFAs also have anti-inflammatory and anti-neoplastic activities in mice, which are mediated by down-regulation of Wnt signaling and induction of apoptosis.69 Patients with CRC show low SCFAs and diminished levels of fecal butyrate and butyrate-producing bacteria.3570
A high-fat diet, which is a common Western-style diet, is closely related to bile acid metabolism. A diet high in saturated fat enhances taurine conjugation and induces the expansion of sulfite-reducing Bilophila wadsworthia. It also exacerbates colitis in IL-10-knockout mice.71 Bile acid levels are controlled by the enterohepatic circulation, and bacterial conjugation in the intestine is a major limiting step. Therefore, alteration of the microbiota composition, so called dysbiosis, increases bacterial conjugation and secondary bile acids via the enterohepatic circulation. Increased secondary bile acids produce reactive oxygen and induce the NF-κB signaling pathway in the intestinal epithelium, resulting in cell damage and cell proliferation.72 Sulfate-reducing bacteria produce hydrogen sulfide as a byproduct of fermentation, which shows genotoxicity.73 B. wadsworthia uses taurine and generates hydrogen sulfide as an end-product. Hydrogen sulfide induces DNA damage and a pro-inflammatory response in the host. In addition to B. wadsworthia, it has been known that various sulfate-reducing bacteria including Desulfovibrio, Desulfomonas, Desulfobacter, and Desulfococcus may be associated with CRC.74 They use lactate, pyruvate, or acetate as an energy source during the reduction of sulfate to hydrogen sulfide.74
In addition, the gut microbiota can influence the mucosal barrier by altering the mucus layer in the intestine. The Western-style diet was shown to alter epithelial permeability and the growth rate of the inner mucus layer in mice.62
PERSPECTIVES
The pathogenesis of CRC is heterogeneous, and has been shown to involve various elements, including genetic susceptibility, host immunity, environmental factors, and the intestinal microbiota (Fig. 1). The intestinal microbiota can influence colorectal carcinogenesis in various ways, by promoting sustained inflammation, impairing host immunity, directly damaging DNA with genotoxins, and altering metabolic activity. Although there is little evidence that the microbiota can induce CRC in humans, results from a F. nucleatum-association study shows strong crosstalk between the intestinal microbiota and the host immune system.
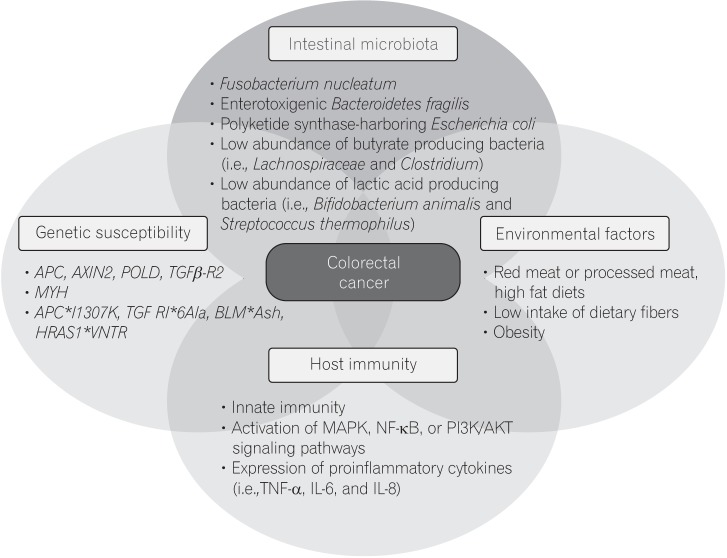
The proposed pathogenesis of colorectal carcinogenesis. MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; PI3K/AKT, phosphoinositide 3-kinase/protein kinase B; TNF-α, tumor necrosis factor α; IL, interleukin.
Most cases of CRC are sporadic, and F. nucleatum may play a role in a late stage of the adenoma-carcinoma sequence. Therefore, it is important to identify the early events in colorectal carcinogenesis. Some bacteria, such as ETBF, induce colon polyps and CRC in the early stage of colorectal carcinogenesis. It is well known that diet affects the intestinal microbiota, and there is growing evidence showing that diet is related to the development of CRC. Long-term intake of a high-fat, low-fiber diet induces dysbiosis and makes the intestinal mucus barrier more vulnerable, leading to changes in the intestinal bacterial composition and stimulation of epithelial cells or the mucosal immune system against intestinal bacteria. In addition, following the change in microbiota composition due to alteration of the mucus layer, reciprocally, the intestinal mucosa becomes even more vulnerable, resulting in DNA damage to the epithelial cells in colorectal adenoma and cancer through the classic adenoma-carcinoma sequence. However, it is uncertain whether dysbiosis precedes the change in epithelial integrity, including the mucus layer, or it happens independently. In addition, both condition have reciprocal effects. Therefore, the conditions associated with dysbiosis may be an initial step in colorectal carcinogenesis. The crosstalk described above will be useful for further promoting the use of colonoscopy, which will affect the prognosis of patients with CRC.
Notes
FINANCIAL SUPPORT: This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2018R1A2B6004475).
CONFLICT OF INTEREST: No potential conflict of interest relevant to this article was reported.
AUTHOR CONTRIBUTIONS: C.H.P. and D.S.H. drafted the article. C.S.E. provided a critical revision of manuscript. All authors approved the final version of the article.